Accurate cost-effective haplotype-resolved sequencing of large targeted genomic regions using HLS-CATCH™ sample prep with TELL-Seq™ library preparation

A single-tube linked-read library prep is used with purified high molecular weight BRCA2 gene targets to demonstrate phased haplotypes of a trio.
Long-range sequencing information is required for haplotype phasing, de novo assembly and detection of structural variation. Long-read sequencing technologies can provide such information, but suffer from high cost, low accuracy, and high DNA input requirements. Universal Sequencing Technology (UST) Corporation has developed a single-tube Transposase Enzyme Linked Long-read Sequencing (TELL-Seq™) library preparation technology, which enables low-cost, high-accuracy, linked-read sequencing on Illumina instruments. TELL-Seq libraries routinely yield linked read maps exceeding 100 Kb with as little as 0.1 ng input material. These characteristics make TELL-Seq an attractive method for sequencing targeted long fragment products produced by Sage’s HLS-CATCH™ method. In these experiments, we demonstrate the benefts of combining UST’s TELL-Seq with HLS-CATCH target enrichment for long range haplotyping at the human BRCA2 locus.
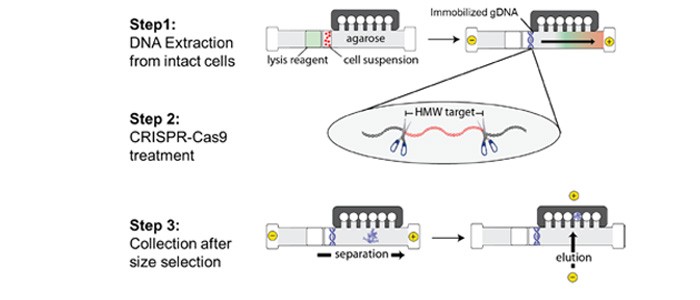
Cultured cells from the Ashkenazi trio of the Genome in a Bottle consortium sample set (NA24395 son, NA24149 father, NA24143 mother) were processed using a SageHLS CATCH work ow (for schematic work ow see Figure 1) optimized for isolation of a 187.5kb target from chr13 containing the entire BRCA2 gene along with 5’ and 3’ anking regions (chr13:32,258,275-32,445,810). Brie y, HMW DNA from 1 million cells was electrophoretically extracted in the sample well of the agarose HLS cassette. After extraction, the DNA was digested (in the HLS cassette) with a Cas9-gRNA mixture that cleaved at the borders of the target fragment. Following Cas9 digestion, the excised target fragments were purifed by automated electrophoretic size selection and electroelution. Eluted target DNA was concentrated and cleaned of contaminating SDS using the Sage Hi-Bead magnetic particle kit (Sage Science). Quantitative PCR was used to locate and quantify the BRCA2 product (Thermo Taqman assay). The HLS-CATCH process yielded 220,000-400,000 copies of the 187kb BRCA2 target from the 1 million cell inputs, with an enrichment of 200-400-fold. Total DNA content of the target fractions ranged from 3-7ng.
Figure 1: HLS-CATCH
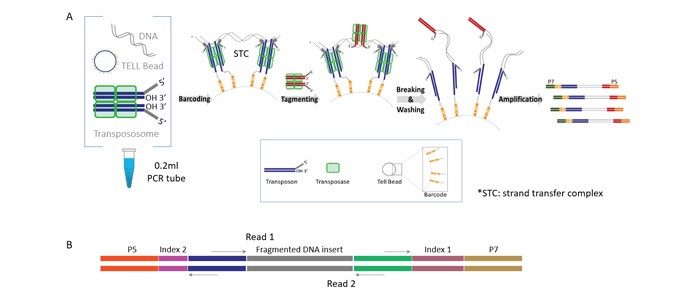
TELL-Seq linked-read library construction (for schematic overview see Figure 2) was carried out with 0.1 ng to 0.2 ng of BRCA2-enriched CATCH product (depending on the enrichment level) using the ultralow input “small-genome” version of the TELL-Seq protocol. Libraries were sequenced on an Illumina MiSeq instrument using 2x150bp PE reads for the father’s cells (NA24149), and 2x71bp PE reads for the mother’s (NA24143) and son’s (NA24385) cells. For the father’s sequencing run, a total of 8.2M cluster reads were obtained, with an on-target fraction of approximately 4%. For the mother’s and son’s sequencing runs, 11.4M and 8.4M cluster reads were obtained from each sample, respectively, with on-target fraction close to 2.5%. The sequencing data were converted to 10X Genomics compatible format and haplotypes were determined using the Long Ranger package (10X Genomics).
Figure 2: TELL-Seq
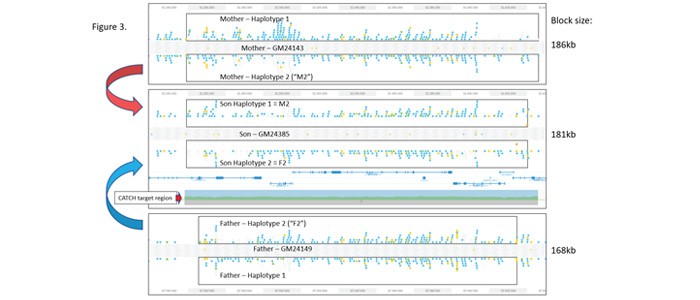
Loupe browser (10X Genomics) views of variants identifed from the three cell lines are shown in Figure 3. In all three TELLSeq data sets, nearly the entire 187kb CATCH target region could be haplotyped in a single phase block. In addition, although the format of the variant displays between the haplotypes is a little variable, the figure shows that haplotype 2 from the mother is identical with haplotype 1 of the son, and haplotype 2 of the father is identical with haplotype 2 of the son.
Figure 3: Trio Data Set
Detailed comparison of the variants in the 6 haplotypes with known GIAB data confirms this arrangement. There were 177 positions that differed from the hg38 reference among the haplotypes. Excluding variants shared by all haplotypes, there were 36 variants that classify the four ~180kb haplotypes within the BRCA2 CATCH target obtained from this family, shown in Figure 4. All 6 haplotypes determined in our CATCH+TELL-Seq experiment agree with the high quality genome sequences for the Ashkenazi family on the Genome in a Bottle website.
Figure 4: Reference Comparison

These data demonstrate that combining Sage’s HLS-CATCH targeted large fragment isolation with ultralow input UST’s TELLSeq sequencing provides a powerful and cost-effective path to accurate long range genomic sequencing and haplotype determination for specific loci without the need for costly whole genome long-read sequencing. Moreover, Sage has found that target recovery of the HLS-CATCH process improves from 25% at 1 million to 50% using low 100,000’s of cells. Since, TELL-Seq can provide high quality data with as little as 0.1 ng of input DNA, the combined CATCH+TELL-Seq workflow may prove very useful for clinical studies with limited input size, such as dissociated biopsy samples.
This content was written by Chris Boles1, Tom Chen2, Peter Chang2 and Long Pham2 and supplied by Sage Science1 and Universal Sequencing Technology Corporation2.