Introducing the next-generation of genetic screening
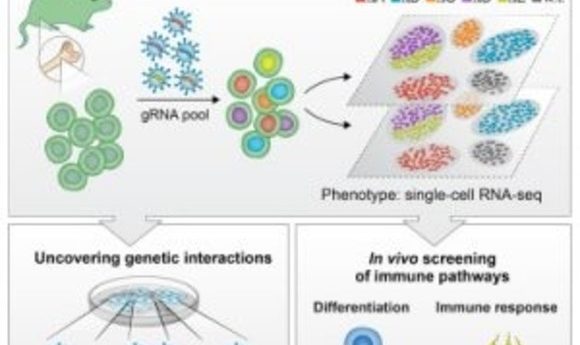
Researchers combine pooled CRISPR screening with single-cell RNA sequencing to enable high-resolution analysis of transcriptional profiles on a large scale.
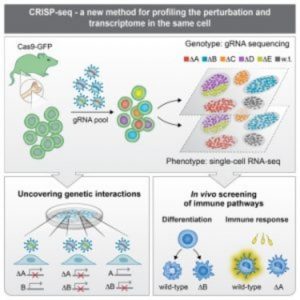
Diagram of CRISPR-seq (4).
Genetic screens are powerful tools for investigating gene function in mammalian cells. Currently, there are two main strategies for associating genetic perturbations with cellular phenotypes. In arrayed screens, which are performed on cells in microwell plates, a single perturbation is delivered into each microwell. Although this approach reveals high-resolution phenotypes based on imaging, transcriptional, or protein-based measurements, it is limited-throughput, costly and labor-intensive (1).
On the other hand, pooled screens allow the rapid survey of many thousands of parallel perturbations in a single sample of cells. Although they are more efficient and scalable, these screens do not reveal a detailed phenotype for each perturbation and generally require secondary validation screens. While pooled CRISPR screens work well for mechanisms that affect cell survival and proliferation, they do not support transcriptome profiling, which is one of the most comprehensive and informative measures of cellular response.
“This approach fails whenever the phenotype of interest is more complex than just counting surviving cells,” said Christoph Bock of the CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences. “In cancer research, you might be interested in upregulation of a key cancer pathway, dedifferentiation to a more immature state, or metastatic potential. These cellular phenotypes are not accessible to classical CRISPR screens, but they can be inferred from a cell’s transcriptome.”
Four recently published studies may have just turned functional genomics on its head by introducing a third alternative that combines the advantages of arrayed and pooled screens. Three independent research teams introduced the methods CROP-seq, CRISP-seq, and Perturb-seq, which use the CRISPR-Cas9 system to generate up to thousands of genetic perturbations in parallel within a single sample, as with traditional pooled screens. But by using single-cell RNA-seq, these approaches enable the perturbation and phenotype of each cell to be measured simultaneously.
These new methods associate rich phenotypic information with each specific perturbation in a plate of cells, enabling single-cell analysis of transcriptional profiles on a large scale. “They establish a novel high-throughput screening paradigm that combines key advantages of pooled and arrayed CRISPR screens, thus contributing to our ability to discover and dissect novel biological mechanisms much more effectively, both in vitro and in vivo,” Bock said.
The Power of Perturb-seq
In two studies published in Cell, a new method called Perturb-seq was introduced by research teams led by Jonathan Weissman of the University of California, San Francisco (UCSF) and Aviv Regev of the Broad Institute of MIT and Harvard. The team at the Broad Institute used this approach to analyze 200,000 cells, focusing on transcription factors regulating dendritic cell responses (2). Perturb-seq accurately identified individual gene targets, gene signatures, and cell states affected by individual perturbations and their genetic interactions.
“Perturb-seq addresses a major gap in current approaches to functional genomics,” said Britt Adamson, co-first author of one of the studies and a postdoctoral scholar in the Weissman lab. “It provides a transformative tool for enabling the rapid and systematic dissection of all of the major transcriptional pathways in the cell.”
This CRISPR/Cas9-based method relies in part on engineered guide RNAs for targeted genome editing. According to the authors, one major bottleneck was developing a cell barcoding strategy for accurately capturing information about which guide RNA was expressed in a given cell and, thus, which gene was being perturbed. It was also a major feat to scale up the number of single cells while accurately assigning perturbation identities to each of them.
“With single-cell data, one is faced with a combination of technical and biological variability overlaid with the perturbation signal that we’d like to detect,” said Atray Dixit, a PhD student at the Broad Institute and co-first author of one of the studies. “It was challenging to come up with methods that could parse out interpretable signals from these sources of variation.”
Meanwhile, the UCSF team simultaneously delivered up to three CRISPR perturbations to dissect the mammalian unfolded protein response, which is triggered by endoplasmic reticulum (ER) stress (3). They used two genome-scale CRISPR interference (CRISPRi) screens to identify hundreds of genes that contribute to ER homeostasis and then applied Perturb-seq to interrogate a diverse subset of these genes with single-cell resolution. These experiments provided insight into how the cellular sensors of ER homeostasis monitor distinct types of stress and highlighted the ability of Perturb-seq to dissect complicated cellular responses.
“Perturb-seq can be used to dissect any biological pathway that has a transcriptional signature or phenotype,” said Thomas Norman, co-first author of the study and a postdoctoral scholar in the Weissman lab. “The complex single-cell phenotypes gathered by Perturb-seq dramatically expand the information that can be gained through screening.”
Expanding Toolkit: CRISP-seq and CROP-seq
In the same issue of Cell, Ido Amit of the Weizmann Institute and his team applied their new method, CRISP-seq, to probe regulatory circuits of innate immunity (4). By sampling tens of thousands of perturbed cells in vitro and in mice, they characterized the regulatory network controlling the differentiation of myeloid cells and their response to pathogens. According to the authors, the findings not only demonstrated CRISP-seq as a versatile approach to probe and infer the wiring of mammalian circuits but also demonstrated its potential for the future engineering of immune cells for therapeutic applications.
Shortly after these three Cell studies were published, Bock and his team described a new method called CRISPR droplet sequencing (CROP-seq) in Nature Methods. Their approach combines four key components: a guide RNA vector that makes individual guide RNAs detectable in single-cell RNA-seq experiments, a high-throughput assay for single-cell RNA-seq, a computational pipeline for assigning single-cell transcriptomes to guide RNAs, and a bioinformatic method for analyzing and interpreting guide RNA–induced transcriptional profiles (5). Compared with Perturb-seq and CRISP-seq, CROP-seq has the key advantage of reading the guide RNA directly, which dramatically simplifies single-cell CRISPR screening with large guide RNA libraries and makes CROP-seq fully compatible with standard cloning protocols for pooled CRISPR screens.
Although this method currently supports only single-cell RNA-seq, single-cell multi-omics protocols are already starting to become available. In future studies, Bock and his team intend to use CROP-seq to explore the potential of epigenome editing in cancer research. One plan is to introduce defined epigenetic defects in normal cells to transform them into cancer cells, with the goal of showing that epigenetics play a causal role in cancer. “This has been fiercely challenged by cancer geneticists,” Bock said.
Conversely, the researchers also plan to reprogram cancer cells into less malignant cells, hoping to identify potential cancer therapies that modulate cell fate rather than killing all cancer cells. “CRISPR epigenome editing and CROP-seq are central to this research because they allow us to introduce targeted epigenetic alterations and to assess their functional consequences in high throughput,” Bock said.
Challenges and Opportunities
The three new methods offer important advantages over previously developed approaches. For one, they provide a one-size-fits-all assay for simultaneously capturing a wide range of phenotypes without relying on biomarkers. They could also be used for applications that were previously difficult to perform in a pooled screen format—namely, combinatorial screens, perturbations in complex cell populations, and screens with rare cells that cannot be cultured. Moreover, these assays exploit the flexibility of the CRISPR-Cas9 system, which can be used for gene silencing (CRISPRi) or activation (CRISPRa), in addition to deletion (1).
From a clinical standpoint, it is possible to envision applying these approaches directly to small biopsy samples containing tens of thousands of cells. “It will be very exciting to apply single-cell CRISPR technology to complex and heterogeneous tissue, including primary tumors where each cell type may respond differently to a drug administered to the tumor as a whole,” Bock said. “With single-cell CRISPR sequencing, we can now test whether different cell types within the same tumor depend on different genes in the way they handle, for example, the challenges of chemotherapy.”
For the time being, several practical hurdles remain. According to Bock, one concern is the high cost of single-cell RNA-seq, which currently limits the scale of screening to about 1000 target genes. But as Dixit pointed out, this cost is expected to decrease over time. “The per-cell cost of performing single-cell RNA-seq has dropped by about 100-fold in the last couple of years and may drop another 10- to 100-fold in the coming years,” Dixit said. “Right now, the cost of doing a genome-wide screen for transcriptional effects in mammalian cells would be on the order of $100,000 to $1 million, depending on how detailed you wanted to get. It’s a lot, but it’s still cheaper with this approach than what it would have been before.”
For their part, Weissman and his team are working on streamlining and scaling their approach. By improving some aspects of the technology, they believe they can increase efficiency and decrease cost. They also plan to continue to develop and refine computational approaches for better understanding the data. “While the technique is still young, there are no intrinsic limitations to expanding the technology even to functional genomics performed at genome-scale,” Weissman said.