Mending muscles with CRISPR
A disorder caused by a mutation in a single gene, Duchenne muscular dystrophy is an attractive candidate for gene therapy; however, the mutated gene happens to be one of the largest in the human genome, making packaging a corrected version into a viral vector impossible. New research now indicates that CRISPR may provide the answer.
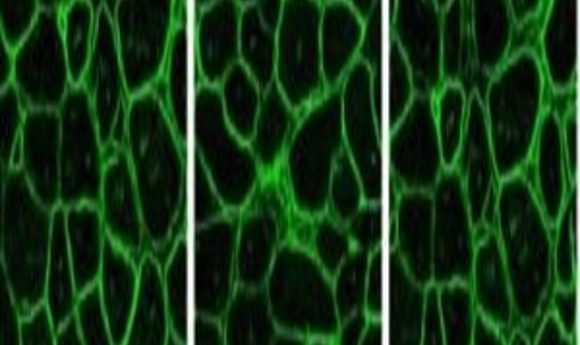
Immunostaining for dystrophin in CRISPR-corrected mouse muscle (5).
Embryonic development is an incredibly complex process where millions of molecular and cellular events come together. Amidst this vast biological landscape, sometimes something as small as a single nucleotide change can seriously alter life.
Such is the case in Duchenne muscular dystrophy (DMD), which affects about one in every 36,000 male children. Because of a mutation in the dystrophin gene, muscle cells become fragile, and muscles waste away, leading to death in the teens or early 20s.
DMD results from a mutation in a single gene, making it an attractive target for gene therapy. But the dystrophin gene is one of the largest human genes—2300kb with 79 exons—making packaging a replacement dystrophin gene into the tiny viral vectors used for traditional gene therapy approaches impossible. In addition, “DMD is a new mutation syndrome, so all patients have slightly different mutations that will require a wide variety of strategies to treat,” said Jeff Chamberlain, a muscular dystrophy researcher from the University of Washington.
Since replacing the gene isn’t an option, researchers have now turned to CRISPR technology to edit it instead. Correcting the dystrophin gene using CRISPR would require cells with active DNA repair machinery, such as those that are dividing. Alternatively, in mature muscle cells, CRISPR can snip out the portion of the gene containing the mutation without subsequently repairing the region, leaving the cell to produce a shortened–but functional–version of dystrophin.
“Gene replacement therapy is easier to apply at the moment, and is to date more effective than gene editing. However, gene editing has great potential, and as more of the limitations are solved I think this approach will become used in patients and eventually gain approval for wider use,” Chamberlain said.
Getting Mice Moving
Researchers have been working on DMD treatments for some time, investigating possibilities such as delivering muscle building stem cells into patients, developing gene therapies to replace faulty dystrophin with a very short version of the gene, using drugs to promote DNA-reading machinery to skip over defective exons, and exploring various gene editing approaches to correct specific mutations. While each line of attack shows some promise, many results in the past have been disappointing to patients and researchers alike.
When CRISPR appeared on the scene, it offered the possibility of packaging a guide RNA and Cas9 DNA-cutting enzyme gene into viral vectors for gene therapy, circumventing the need to package portions of the cumbersome dystrophin gene. In 2014, Eric Olson’s team at the University of Texas Southwestern Medical Center used CRISPR to correct a dystrophin mutation in the germline of a DMD mouse model. The researchers recorded a wide range in the percentage of cells carrying the modified gene in mouse pups born to treated parents, but germline editing prevented muscles from degenerating even in mice with few corrected cells (1).
Germline editing isn’t feasible for humans, but the promising effects seen in the health of the mice encouraged researchers to pursue this line of research, altering their strategy to accommodate mature muscle cells that are no longer dividing. Olson’s team followed up in 2016 by using an adeno-associated virus-9 vector (AAV) to deliver CRISPR elements to DMD mouse models, deleting a faulty exon and removing a frameshift mutation. They tested systemic and muscle-specific injection of the vector on different days after delivery of the pups and found that each method restored dystrophin protein expression and enhanced cardiac and skeletal muscle performance (2).
In that same issue of Science, Charles Gersbach’s team from Duke University (3) and Amy Wagers’ team at Harvard University (4) also used CRISPR to delete the same dystrophin exon and reported improved muscle function and biochemistry. Importantly, Wagers also showed that this approach corrected dystrophin in muscle stem cells, meaning that the effects of the treatment were likely to continue long-term.
“[These papers] all targeted the same, single mutation using essentially the same strategy,” Chamberlain said. “We wanted to ask whether other types of mutations could be targeted, especially ones that were not the simplest case scenario.”
Expanding Possibilities
Chamberlain’s team recently decided to test the potential for CRISPR to treat a different mouse model. In this case, correcting the mutation required deleting a large genomic region or directly fixing a specific mutation.
“The only current method to achieve gene editing in muscles body wide requires delivery of the CRISPR/Cas9 machinery to muscles using AAV vectors,” explained Chamberlain. “However, these vectors target cells and tissues all over the body, especially liver. As a result, the method delivers large amounts of Cas9 nuclease to muscle and non-muscle cells, allowing for long-term target and off target gene editing in many non-muscle cells, including dividing cells that could acquire uncontrolled growth.”
Chamberlain’s team employed a muscle-specific regulatory cassette to contain Cas9 expression to non-dividing muscle cells and devised two gene editing strategies. In the first, they removed exons 52 and 53, which encode non-essential portions of the dystrophin protein; the second approach directly corrected a premature stop codon in exon 53 to restore production of the full-length protein.
The team recently reported in Nature Communications that these approaches induced muscle-specific functional dystrophin expression that improved both skeletal and cardiac function in a mouse model of DMD (5). “Our results suggest that gene editing could be applied to the majority of mutations that cause DMD, although probably not all,” Chamberlain reported.
As researchers inch ever closer to an effective treatment for DMD, several important tasks remain before testing CRISPR gene editing approaches in larger animals and humans. “The efficiency needs to be improved to achieve a therapeutically-relevant level of gene correction,” Chamberlain said. “The method also needs to be adapted for more mutation types. Off-target editing should be reduced for safety, and finally it will be important to find ways to keep Cas9 nuclease activity minimal after the initial wave of editing occurs.”
Chamberlain and his team are currently working on these tasks, as well as exploring different mutations in dystrophin that may lend themselves to correction by gene editing and alternate systems for testing possible therapies.