Single-cell methylomes
DNA methylation patterns in thousands of single cells reveal expanded neuronal diversity in the human brain.
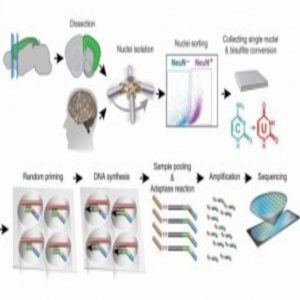
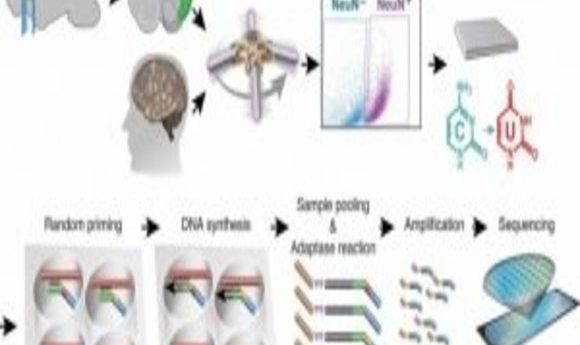
High-throughput single-nucleus methylome sequencing (snmC-seq) of mouse and human frontal cortex neurons (1).
Sequencing mRNA transcripts from single cells or nuclei enables researchers to identify cell types with unique transcriptional profiles in the brain. But these methods are restricted to RNA signatures, which are influenced by the environment. On the other hand, DNA methylation is an epigenetic mark that distinguishes cell types and marks regulatory elements, while remaining stable across individuals and over the life span.
“Many groups use single-cell RNA-seq to study cell-type diversity,” said Chongyuan Luo from the Salk Institute for Biological Studies, first author of a new paper in Science. “We have demonstrated that neuron types can be classified with comparable resolution using DNA methylation profiles.”
To complete this study, Luo and his collaborators developed a protocol for single-nucleus methylcytosine sequencing (snmC-seq) and applied it to neurons from the frontal cortex of the mouse brain and the human brain. The researchers generated methylomes from more than 6000 mouse and human neurons, covering approximately 5% of the genome per cell.
“This is the first time anyone has measured the DNA methylation pattern of thousands of single cells,” said study author Eran Mukamel, assistant professor of Cognitive Science at the University of California, San Diego.
Applying a uniform experimental and computational pipeline to mouse and human neurons enabled an unbiased comparison of neuronal epigenomic diversity in the two species. The team identified 16 mouse and 21 human neuronal subpopulations in the frontal cortex. “We are excited by the finding that the human cortex shows more expanded neuronal diversity than the mouse cortex,” Luo said.
“The research helps to more clearly define the similarities and differences between neuron types, especially in terms of their epigenetic regulation,” Mukamel said. But further anatomical, physiological, and functional experiments are needed to characterize the DNA methylation–based neuronal populations defined in the study and to enable identification of cell types that cause human neurological diseases. “Our next goals are to apply this technique across different brain regions, to fill out the picture of brain cell types. We also want to know how different experiences and chemical or environmental exposures impact the neuronal epigenome, and this technique could be helpful with that.”