Pinpointing pain: researchers discover protein subunits that respond to opioids
A discovery about voltage-gated calcium channels and opioids could help develop more targeted drugs for pain.
Opioids are a key drug used for treating pain; however, we still don’t know exactly how they work. Now, researchers from Linköping University (Sweden) have uncovered the exact subunits of calcium channels that respond to opioids. The finding could help develop more targeted pain drugs with fewer side effects.
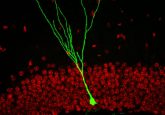
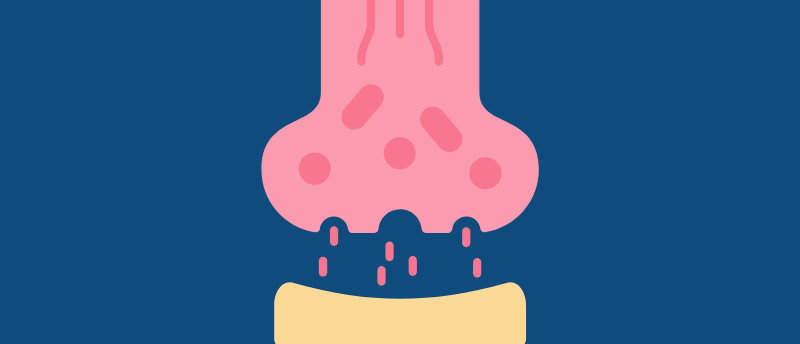
Pain is conducted through our nervous system mainly as electrical signals. However, between neurons, the electrical signal must be converted into a biochemical one. Voltage-gated calcium channels are used for this. When an electrical signal reaches the end of a neuron, voltage-gated calcium channels open, allowing an influx of calcium, which triggers the release of neurotransmitters. This signal is received by the next neuron, which converts it back into electricity, and so on.
CaV2.2 is a specific type of calcium channel located at presynaptic terminals of sensory neurons that play a key role in pain signaling and are more active in chronic pain. Drugs that target CaV2.2 exist; however, they have challenges. For example, opioids, like morphine and heroin, are a drug class that targets CaV2.2 channels. Opioids exploit a natural mechanism to decrease the ability of CaV2.2 to respond to pain signaling. While opioids are effective at reducing pain, they’re also highly addictive.
It has been established that opioids trigger the release of G proteins, which bind to CaV2.2 channels and make them ‘reluctant’ to open. However, the mechanism by which this happens is unknown.
 Untangling the role of TYK2 in Alzheimer’s disease tau pathology
Untangling the role of TYK2 in Alzheimer’s disease tau pathology
TYK2’s role in the development of intracellular tau neurofibrillary tangles associated with neurodegeneration has been discovered.
CaV2.2 has four voltage-sensor domains (VSDs), VSD I-IV. These VSDs detect electrical nerve impulses, moving and opening the channel when the voltage is high enough. To investigate how G proteins regulate this process, the researchers utilized a hybrid electrophysiological and optical approach, called voltage-clamp fluorometry, which provides information about the conformational changes of voltage-gated proteins.
They found that the individual VSDs respond differently to voltage and modulation by proteins. Under normal conditions of pain, VSD-I and -IV activations resemble pore opening, VSD III activation is hyperpolarized and VSD II does not respond to voltage changes. In the presence of the G protein complex Gβγ, which is the result of G-protein-coupled receptor signaling in the presynaptic terminal, pore opening and VSD I are strongly inhibited, VSD IV is modestly inhibited and VSD III is not inhibited.
The findings suggest that specifically inhibiting VSDs I and IV could be a way of regulating CaV2.2 channels, and therefore pain. “Our finding points to a very specific part of the large calcium channel that next-generation drugs can target to provide pain relief in a similar way to opioids,” commented corresponding author Antonios Pantazis. “Instead of blocking the calcium channel completely, which is a less refined method, future drugs can be designed to fine-tune calcium channel activity in pain signaling.”