Real vs. reference genomes
Researchers rely on bacterial reference genomes, but how accurate are they? A new study raises concerns.
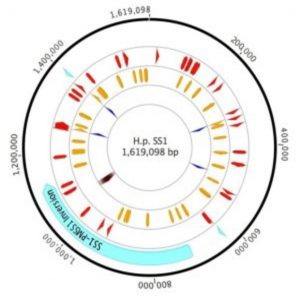
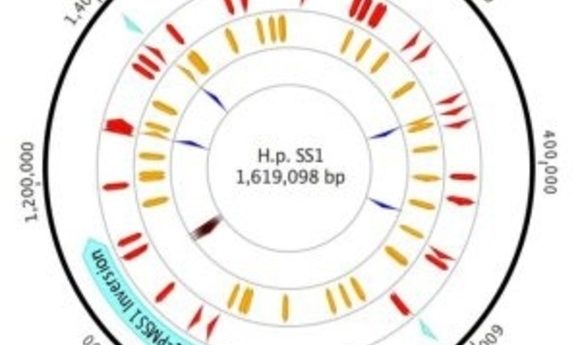
Plot of H. Pylori strain SS1 genome highlighting sites of variation (1).
It is not news that genomes vary among individuals within a species—yet researchers often rely on published reference genomes to guide their work. Reference genomes represent consensus assemblies, usually generated from single colonies of a given strain, that attempt to account for the most common variants. But how accurate are they? Can they, for example, capture internal genomic rearrangements?
Now, in the journal mBio, a team from the University of California, Santa Cruz and other institutions report next-generation deep sequence analyses that address these questions for the bacterium Helicobacter pylori.
“Everybody is aware that bacteria are not static, but investigating genomic variation within lab stocks is both expensive and time-consuming.” said Jenny Draper , the lead author of the study.
Draper and her colleagues compared inter- and intra-genomic variability from typical laboratory working stocks of two reference strains of H. pylori: PMSS1, a parental strain isolated from a human gastric ulcer patient, and SS1, a PMSS1 descendant that has been passed through mice.
While the SS1 genome was fairly typical for H pylori, the researchers observed 46 mutations relative to PMSS1, including some in genes relevant to pathogenesis and mouse adaptation. However, they also found significant variation within the SS1 working stock population. For example, they observed a transposon moving within the genome, dynamic variation in cagA (a virulence gene) copy number, and nearly 60 SNPs.
“I think what we are reporting is not unexpected…H. pylori is highly mutable, perhaps because of evolutionary adaptations to host selective pressures,” Draper said. However, she was surprised to see differences in the copy numbers and tandem duplication of cagA. “It’s the most important gene for pathogenesis and very well studied, but nobody had reported this duplication.”
Draper compares the movement of the transposon in SS1 (another unexpected finding) to Schrödinger’s cat. “It was present and it was not present at the same time! For 3 of 4 observed insertion sites, the transposon was both there and not there—simultaneously—in the population. About 10% of the time, this gene has a transposon in the middle of it; the rest of the time it’s fine.”
Sequencing H. pylori at such high resolution took years, but Draper said the most challenging part was assembling the variable genome.
Despite the complexity of the project, Draper’s take home message is a simple one: “If what you are looking at is a population of bacteria, it may be different from what you think it is. But if you don’t work with a highly mutable organism and don’t do deep sequencing, you probably won’t see it.”